本文是一篇计算机论文,计算机的应用在中国越来越普遍,改革开放以后,中国计算机用户的数量不断攀升,应用水平不断提高,特别是互联网、通信、多媒体等领域的应用取得了不错的成绩。(以上内容来自百度百科)今天为大家推荐一篇计算机论文,供大家参考。
第 1 章 绪 论
1.1 课题研究背景
随着各种生物芯片技术及高通量测序技术的日益发展,各种组学数据,如基因组,转录组,蛋白质组及代谢组学数据源源不断地涌现。这些组学数据在生命科学大数据中占据重要地位,是除了生物图像数据外占有比例最大的数据。例如,在 RefSeq数据库中有超过 2 亿的 DNA 序列和超过 8 千万的蛋白质序列;在 GEO数据库中有超过 10 亿的基因表达谱数据。面对大量组学数据不断地积累,传统的实验生物学已不能满足现代研究需求,而以计算机科学为基础,利用数学以及生物学各种工具的交叉学科——生物信息学,在该研究领域的重要作用日益凸显。结合已有的生命科学机理知识,生物信息学方法可以充分利用海量组学数据构建计算模型,有助于突破传统的生命科学的实验研究方法的局限,从而有望发掘蕴含在海量组学数据中的生命本质规律。在这些生物组学数据中,蛋白质组是生物功能的执行者,是最直接与生命功能相关的。前体蛋白是没有活性的,通常要经过一系列的加工才能成为具有一定功能的成熟蛋白。蛋白质翻译后修饰(PTM:Post-Translational Modification)是其中的一种蛋白质加工类型,其通常是指一个功能集团(functional group)或者小分子量的蛋白与蛋白质氨基酸序列的特定位置的共价结合,例如磷酸化,乙酰化,甲基化及泛素化等。PTM 是增加生物多样性的关键机制[1]。有研究表明,真核生物细胞中存在着各种各样的蛋白质修饰过程,已经确定的 PTM 种类就高达 400 多种[2],他们几乎影响细胞生物学和发病机制的方方面面,但目前的研究仍有大约 70%的 PTM 过程无法解释。PTM 研究的一个关键问题是全面及准确地识别蛋白质序列上的 PTM 位点。虽然通过高精度的质谱检测就可以检测到特定的 PTM 位点,但该方法价格昂贵、耗时,不适合大规模的检测,更不可能进行全蛋白质组级别的识别。相较于常规实验方法,生物信息方法的计算识别为蛋白质组的注释和大规模实验设计提供了一种替代策略,具有速度快,成本低等优点。
.........
1.2 课题研究意义
蛋白质翻译后修饰(Post-Translational Modification,PTM)在生命活动中具有十分重要的作用。PTM 相关问题研究的重要性已经被很多诺贝尔奖认可:例如,EdmondH.Fischer 和 Edwin G . Krebs 通过发现可逆蛋白磷酸化过程被授予 1992 年诺贝尔奖[24];又如 Leland H. Hartwell,Tim Hunt 和 Paul M. Nurse 于 2001 年被授予诺贝尔奖,以表彰他们发现的关键细胞周期调节机制,包括与磷酸化过程相协调的细胞周期依赖的激酶和细胞周期过程[25]; 以及 Aaron Ciechanover,Avram Hershko 和 Irwin Rose 等人通过发现泛素化介导的蛋白质降解机制而被授予的 2004 年的诺贝尔奖[26]。研究表明 PTM 过程几乎影响正常细胞生物学和发病机制的方方面面,包括细胞分化[27],蛋白质降解[28],信号和调节过程[29],基因表达调控[30],以及蛋白质-蛋白质相互作用[31]等关键细胞过程中都发挥着重要作用[32, 33]。PTMs 的异常还与疾病和癌症密切相关,而参与 PTMs 的各种调节酶已成为药物靶点。PTM 的相关问题已成为当今蛋白质组学研究中的重要课题,而对于 PTM 位点的全面及准确的识别是该问题研究的关键,同时也是 PTM 问题的研究热点与难点。研究深度学习方法在蛋白质翻译后修饰位点预测问题中的应用具有双重意义。从生物数据分析的角度来说,首先,深度学习方法提供了一种新的分析手段。传统的机器学习方法在解决 PTM 位点预测问题时通常都依赖于预先对蛋白质序列数据的特征提取过程,然后再经由一种机器学习方法对提取的特征进行训练和预测。这种特征提取的过程被称为“特征工程”,其往往受限于研究者已有的背景知识,需事先知道与各种 PTM 位点的预测有关的特征。这就大大地限制了机器学习算法的应用,因为只有少数研究比较透彻的 PTM 才可能会找到合适的特征,而对于大多数的 PTM,该类方法难以操作,并可能会导致有偏差的特征。与传统的机器学习方法相比,深度学习方法接受原始的蛋白质序列进行分析,可在训练过程中自动地生成适合于分类的特征表示,因此是一种更有潜力的解决方案。由于是数据驱动的方式,从而避免了人为干预导致的偏差。第二,深度学习是天然的增量学习模型。增量学习(IncrementalLearning)是指一个学习系统能不断地从新样本中学习新的知识,并能保存已经学习到的知识[34]。
...........
第 2 章 深度学习的相关知识
深度学习方法并不是单一的算法,而是一类学习框架,包含很多具体实现不同的算法。深度学习中最基本的方法是多层神经网络,它在原有的浅层神经网络结构的基础上增加了隐层的个数,从而扩展了神经网络对更加复杂的数据处理的能力。除了多层神经网络,常见的深度学习方法还有深度卷积网络以及循环神经网络。根据具体问题的不同,这些网络有不同的适用范围。其中,深度卷积网络在处理图像,视频,语音和音频方面取得了突破性进展[10, 11, 17, 18],而循环神经网络则极大地提高了对文本和语音等连续数据的分析性能[19, 20]。本章就本文的研究问题——蛋白质翻译后修饰位点预测中涉及到的深度学习模型,包括卷积神经网络、循环神经网络,以及 Hinton 于 2017 年提出的新型深度学习模型胶囊网络进行了介绍,并对一些相关的深度学习技术与深度学习的网络训练方法进行了介绍。
2.1 卷积神经网络
卷积神经网络(Convolutional Neural Network,CNN),于 1989 年由 LeCun 首次提出,是一种用于处理具有网络结构的数据的特殊的神经网络[58]。1998 年,LeCun 将一种称为 LeNet-5 的多层 CNN 网络用于手写数字识别,由此确立了 CNN 网络的现代结构[59]。在 2012 年的 ImageNet 竞赛中[7],由 Alex Krizhevsky 提出的深度卷积神经网络AlexNet 展现了前所未有的性能,从此 CNN 网络受到了广泛关注。卷积神经网络已经在图像识别、分割、分类,以及语音识别等多个领域广泛应用,并获得了突破性进展。卷积神经网络的关键在于网络中至少有一层使用了卷积(convolution)运算。卷积运算是一种特殊的线性运算。在卷积神经网络中定义的卷积运算与其他领域定义的卷积运算不完全一致,本节以一种在很多机器学习库中实现的卷积运算进行介绍。
........
2.2 循环神经网络
循环神经网络(Recurrent Neural Network,RNN)是一种更适合于处理序列数据的神经网络。传统的前馈神经网络模型是从输入层到隐含层再到输出层的全连接网络,其中每个连接具有独立的参数,需要分别对其进行学习。面向具有序列特征的数据,传统的前馈神经网络是低效的,因为它对序列的每个位置单独学习,而忽视了序列上元素间具有的前后依赖关系。而循环神经网络可以在时间维度上共享权重,因此可以考虑序列中相邻位置的关系。基本的 RNN 网络的结构可以表示成图 2.2 的形式,即隐藏层的输入不仅仅来源于当前时刻的输入层,还包括上一时刻的隐藏层的输出。
.........
第 3 章 基于深度学习的通用磷酸化位点预测方法.....27
3.1 通用磷酸化位点预测问题定义....... 27
3.1.1 磷酸化生物背景介绍..... 27
3.1.2 通用磷酸化位点预测问题定义........... 28
3.1.3 以 Musite 为代表的其他磷酸化位点预测方法介绍 .......... 29
3.2 面向通用磷酸化位点预测问题的深度学习框架 ........... 30
3.3 针对不平衡训练数据的学习策略.............. 38
3.4 实验验证与结果......... 40
3.5 本章结论与讨论 .......... 49
第 4 章 基于迁移学习的激酶特异性磷酸化位点预测方法...............51
4.1 激酶特异性磷酸化位点预测问题定义...... 51
4.2 面向激酶特异性磷酸化位点预测问题的深度学习框架 .......... 52
4.3 实验验证与结果 .......... 54
4.4 MUSITEDEEP 工具包的介绍............. 61
4.5 本章结论与讨论 .......... 62
第 5 章 蛋白质翻译后修饰问题中的两种学习策略.....65
5.1 物种间串行化训练策略........ 65
5.1.1 串行化训练方法............... 65
5.1.2 实验结果..... 67
5.2 模型并行化训练策略 ............. 68
5.3 本章结论与讨论......... 71
第 5 章 蛋白质翻译后修饰问题中的两种学习策略#p#分页标题#e#
本章以磷酸化为例介绍蛋白质翻译后修饰问题中两种可行的学习策略:一种是训练数据串行化的训练策略;另外一种是训练模型并行化的训练策略。在本文第四章中介绍的用于激酶特异性磷酸化位点预测问题的迁移学习方法就属于一种训练数据串行化的学习策略,即先以通用磷酸化训练样本训练基础模型,再以激酶特异性训练样本在基础模型的基础上训练激酶特异性磷酸化位点预测模型。第四章中的串行策略考虑的是 PTM间的层次关系,并只针对人类物种训练预测模型,而本章的串行化训练策略引入了物种间的关系。本章的并行化训练策略同样考虑了 PTM 间的关系,但是却以另外一种模型实现。本章分为三个部分,首先对物种间串行化训练策略及实验结果进行介绍,然后介绍模型并行化训练策略及实验结果,最后是本章结论与讨论。
5.1 物种间串行化训练策略
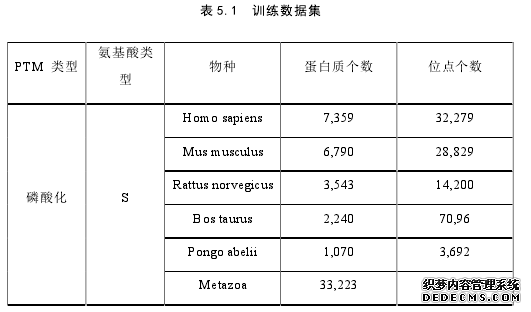
以磷酸化为例,同属于一个科目下的物种由于蛋白质序列上的相似性,因此享有一些共同的激酶,同时也可能有物种特异性的激酶。有一些计算方法,将所有的物种混合在一起对模型进行训练,该类方法的代表有 ModPred[3]。而以 SVM 为基础的磷酸化位点预测方法 Musite 却证明了物种特异性的模型针对同一物种的预测结果要优于跨物种的模型以及混合物种的模型[51]。本文的物种间串行化训练策略基于的假设是物种混合模型可以识别出物种的共性特征,物种特异性模型可以识别出物种特有的特征,而物种混合模型可以帮助物种特异性模型的训练。基于这个假设,本文提出的物种间串行化训练策略的具体做法是,先用混合物种的训练数据预先训练一个基础模型(如图 5.1 左),再利用特异物种的训练数据及迁移学习的思想,在基础模型的基础上迁移学习得到物种特异性模型(如图 5.1 右)。这样,既可以利用混合物种模型中更多的注释数据学习到物种间通用的特征,又可以针对特异的物种对模型细化。本节以通用磷酸化位点预测问题为例,介绍该串行化训练方法。
..........
总结
MusiteDeep 是本文提出的首个用于通用及激酶特异性磷酸化位点预测问题的深度学习框架。与传统的基于统计或机器学习的磷酸化位点的预测方法不同,MusiteDeep 不需要人工的特征选择过程,而是从原始的蛋白质序列着手,通过多层卷积网络及一个双方向的注意力机制自动地生成利于磷酸化位点预测的蛋白质序列表征。MusiteDeep 在通用磷酸化位点预测的 ROC 和 PRC 曲线及曲线下面积等方面的表现都优于其他方法,特别是在平均精度(PRC 曲线下面积)上,高出其他方法最好成绩的 50%。为 MusiteDeep设计的双方向的注意力机制可以分别定量衡量序列维度和特征维度两个方向上元素的贡献,并通过融合这两个维度的信息为给定的蛋白质片段生成具有一定生物解释的特征表示。为解决 PTM 位点预测问题中的正负样本不平衡问题,MusiteDeep 成功引入基于Bootstrapping 的深度学习训练策略,使得学习过程的每次迭代都在比较平衡的训练集合上进行。通过与多种学习策略的比较,MusiteDeep 的双方向的注意力机制与Bootstrapping 训练策略展现了更优的预测性能。本课题的研究还对其他成功应用于基因组学的深度学习框架在通用磷酸化位点预测问题的应用进行了探索,并展示了相应模型的预测效果。另外,利用迁移学习的思想,MusiteDeep 框架缓解了深度学习方法在激酶特异性磷酸化位点预测的小样本学习问题,达到了与现有方法同等或者更好的表现。MusiteDeep 方法最终以开源代码工具包的形式发布,为生物及生物信息科研人员提供了一个可以自定义的 PTM 位点训练和预测的工具:(1)用户可直接使用 MusiteDeep 预先训练的通用或激酶特异性磷酸化位点预测模型对自己的蛋白质数据进行预测;或者(2)用户提供自定义的其他 PTM 位点训练集,使用 MusiteDeep 进行自定义训练,再对测试集进行预测。
..........
参考文献(略)

