本文是一篇计算机论文,计算机的应用领域从最初的军事科研应用扩展到社会的各个领域,已形成了规模巨大的计算机产业,带动了全球范围的技术进步,由此引发了深刻的社会变革,计算机已遍及一般学校、企事业单位,进入寻常百姓家,成为信息社会中必不可少的工具。(以上内容来自百度百科)今天为大家推荐一篇计算机论文,供大家参考。
第一章 绪 论
1.1 研究的背景和意义
胰腺癌是在全世界范围内发病逐年增加的恶性程度很高的消化系统肿瘤,可以说是五大死亡癌症[1-4]。胰腺癌的临床表现与癌症的部位有很大关系,多发生转移,就更难以分析及其诊断,尤其难于治疗。针对胰腺癌的靶向性治疗一般处于临床前期,难以形成有效的靶向性治疗。典型发病致死速度快,一半以上病人死于诊断后 5 个月内可以说是“癌症之王”。肾上腺皮质癌是一种侵袭性的癌症,其年发病率约为 1-2/100 万。有的肾上腺皮质癌,所有的临床和部分免疫组织化学特征都是播散性未分化的胰腺癌,并且“不小心”被当作胰腺癌进行治疗[5-8],耽误了相关治疗,神经胶质瘤与胰腺癌临床表达症状基本一致所以急需一些有效的方法区分,并有针对性靶向治疗。神经胶质瘤也叫胶质瘤,是比较常见的原发性系统疾病[9-12]。引发神经性胶质瘤的原因并不明确,按照胶质瘤分类,可以分为星型细胞瘤、少支胶质瘤、混合类胶质瘤等多种种类。选取这三种癌症为研究对象是首先这三种癌症都属于遗传性癌症[13]。胰腺癌患者已确定有一定机会发生垂直关系,一般为常染色体遗传[14]。肾上腺皮质癌发病原因尚不明确,但是确定 SF1 在胚胎期就参与肾上腺发展,遗传性 SF1 缺失将导致肾上腺皮质癌的发生[15]。神经胶质瘤为原发性中枢神经系统肿瘤,一般为遗传高危因素及环境致癌相互作用引起[16]。其次在临床表现很难区分,有一定临床相似性,对临床诊断有一定指导作用。最后了解了三种癌症的 miRNA 和基因的异常表达网络,可以根据网络通路、环路,研究共有通路及个性通路对靶向性治疗提供一部分参考作用。在临床上神经胶质瘤与其他两种很难区分,临床表达类似,对确诊后进一步治疗提供了很大的障碍。本文的出发点也是从这出发,找出异常表达通路[17-19],结合已经证实的通路,分析 miRNA、基因的表达形式,及网络构造,找出各自及相关特点,以此为根据可以显著区分三种癌症,为临床三种癌症的区分提出相关靶向性治疗提供指导[20]及遗传性分析为早期预防提供依据[21-22]。
.........
1.2 基因表达的研究背景
转录因子(TFs)是一群能与基因 5`端上游特定序列专一性组合,能与特异核苷酸序列上的蛋白质结合,活化后从胞质转位至胞核通过[56]。如图 1.3 可以看到人类金属硫基因调节区。miRNA 是一类广泛存在于真核生物中的内源性非编码 RNA,长度在 19~24nt 之间,在转录后水平上通过调节其靶基因行使功能[60-61]。靶基因也叫目的基因,能相应生物体产生相应的临床表现。自从 2003 年第一个 miRNA 靶基因软件问世,预测靶基因软件可以说是层出不穷。最具代表性的 targetcscan、miRBase 等数据库。一个 miRNA 往往对应着多个靶基因,形成了如图 1.5 中对应关系。近几年来,研究者对人体 miRNA 和基因研究产生的海量数据,生物信息学研究重点已经转移到如何解释这些数据并从中挖掘潜在的知识。利用生物信息学对 miRNA 及靶基因,miRNA 转录因子之间的关系,进行解释及区分相关病症。三角环形结构转 TF 调控miRNA,miRNA调控protein target,miRNA与其宿主基因间关系仍是研究的难点[62-66]。
........
第二章 基于大数据挖掘三种癌症异常表达数据
2.1 概述
大数据挖掘是互联网兴起后,产生大量互联网数据,这些数据可以广泛应用,在迫切需要把这些大数据转化为有用知识的产物。从图 2.1 我们可以发现从 2001 年-2014年 miRNA 文章数据可以说呈几何数量增长,如何把这些有用的数据去伪存真进行分析是今年来一个迫切需要解决的问题。本文挖掘 miRNA、gene、host gene、target gene、tfs 等相关数据,重构相关数据关系,建立数学模型,利用异常表达网络、聚类热图等大数据方式对这些数据进行分析。miRNA 通过结合 RISC 并作用 mRNA 的 3’UTR 上,降解其靶 miRNA 或抑制其靶 miRNA的翻译,广泛参与与细胞的增值分化、发育凋亡等多种生物学过程,以此产生多种疾病。通过研究这个 miRNA 的靶基因,把他们归纳到一起形成数据库,我们会选取数据比较准确的几种数据库进行靶基因预测如表 2.1,我们可以发现多个靶基因的数据库,原理都是类似。miR-17,miR-21,mir155 等都是比较早确定与胰腺癌与肾上腺皮质癌的 miRNA,如mir17 确定为胰腺癌作用中促进细胞增殖[110],miR-21,miR-155 作为典型原癌基因,miR155基因的过度表达是由BIC基因第 241到262区间的编码miRNA,miR-155基因的过度表达激活了癌基因 MYC。以胰腺癌为例图 2.2 中以 24 例胰腺癌为蓝本,平均 63 个基因改变,多为点突变。
.........
2.2 大数据挖掘
以往的相关联的 miRNA 的都是传统的数据挖掘的方法建立,没有利用大数据方法。随着互联网数据不断深入 miRNA 参考文献及数据越来越多挖掘消耗的时间较大,也可能导致数据量搜索覆盖不足,而导致分析失败。利用互联网大数据方法进行研究已经刻不容缓。根据数据库权威性及覆盖面等,我们选取部分数据库,根据数据库特点,建立相关搜集策略,形成数据挖掘模型,对数据进行挖掘,根据数据库结构不同采取不同的策略,如对某些正如某些蛋白质编码基因可能在同一个细胞通路中有协同作用一样,KEGG等数据库为可视化数据库,则需要采用文本挖掘的方法与人工收集相结合的方法。利用贝叶斯聚类分析方法对文本处理过程分析的语料(text corpus),就是通过各大数据库查找的出版物,如(PubMed、NCBI 数据库等)收集的出版物的题目、关键词,高频词等,根据这些语料建立半结构化的文本库(text database)。本文文本挖掘利用Python、R 语言,涉及文本挖掘相关的 R 程序包:tm、lsa、RTextTools、textcat、corpora、zipfR、maxent、TextRegression、wordcloud。本文主要采用的 tmR 包(英文分词)jiebaR包(中文分词),klaR 包(分类器)等。从已有数据库里挖掘与三种癌症和 miRNA 有关的数据,并建立数据集合。miRNA 信息的出版物数量已经多到用人工收集信息方法无法完成,开发计算机方法从文本中提取生物实体(例如基因和疾病)之间的关系已作出重大的突破[ 111-114]。有三种主要的研究方法:基于共通方法、基于规则和机器学习的方法。基于共通方法[115-117],在同一句子或段落之间的同现关系被认为表示一种关系,如在 wang j 等建立的转录因子 microRNA 调控数据库[118]。也可以用其他证据来加强联系或丰富关系。在基于规则的方法、语言学和/或生物知识的编码规则,可以作为汽车自动程序,从文本中提取关系实现,如 Smoot ME 等人利用数据集成化,把这些知识包括纯文本/句法,具体表明生物关系和滤波算法,并可以转化为确定性的步骤提取关系[ 119 ] 。机器学习方法将关系抽取看作一个分类问题,并尝试应用机器学习算法[120]或朴素贝叶斯分类器[121]来提取关系。B 在对预测建模和分析的基本方法进行了深入的讨论。虽然这种类型的概述已经尝试过,这种方法提供在该领域的初学者和专家都喜欢玩的一步的方法。在研究的任务,如蛋白质相互作用关系抽取[122],机器学习方法已被广泛应用。
........
第三章 数据处理及通路构建....37
3.1 数据整理及补充.....37
3.2 数据处理集合.........38
3.3 异常表达网络的建立.......40
3.4 本章小结....... 44
第四章 基于基因和 microRNA 的三种癌症异常网络研究......45
4.1 引言..... 45
4.2 胰腺癌异常表达网络.......46
4.3 肾上腺皮质癌异常表达网络.....49
4.4 神经胶质瘤异常表达网络.........51
4.5 三种癌症的异常表达网络异同对比分析.....53
4.5.1 引言....... 53
4.5.2 胰腺癌与肾上腺皮质癌网络通路对比..........54
4.5.2 胰腺癌与神经胶质瘤网络通路对比....56
4.5.4 三种癌症的异常表达网络通路对比....63
4.6 本章小结........ 66
第五章 结论和展望..........67
5.1 工作总结....... 67
5.2 不足及下一步展望...........69
第四章 基于基因和 microRNA 的三种癌症异常网络研究
4.1 引言
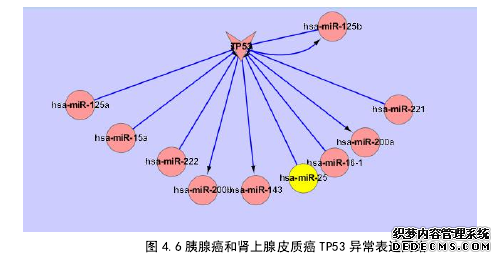
利用大数据方法收集的各种数据库中胰腺癌、肾上腺皮质癌、神经胶质瘤的异常表达 gene、miRNA、靶基因、宿主基因等多种数据。通过文本挖掘及数据整理、数据补充等方面,建立了 miRNA 与 gene 的对应关系。数据挖掘我们利用了文本挖掘和人工挖掘相结合的方法了 pubmed 数据库、NCBI GENE 数据库、NCBI SNP 数据库等,搜集了三种癌症多达 15 万条数据,经过筛选,过滤有用有关 miRNA 和基因数据 1 万多条。同时我们也运用了人工收集的方法,对可视化数据库 KEGG、TCGA 等数据库进行人工挖掘,利用工具获得相关数据。利用随机漫步信任路径算法,利用癌症相关性原理,选了前 5 名最相关的 miRNA 数据,我们补充了胰腺癌 1 个 miRNA、肾上腺皮质癌 2 个 miRNA、急性白病 2 个 miRNA 数据,有利于我们更好的构造异常表达数据库。为了更好利用已经采集的数据,具体分析“为什么发生”,建立数据分析模型。本文主要采用了两种方式:第一种方法是利用 Cytosape 软件构建了三种癌症关于 miRNA、靶基因、转录因子的实验证实调控网络,建立可视化分析网络图,直观的对数据进行分析;第二种方法利用类聚观点利用 miRNA 和基因对应关系采用类聚热图方式,体现三种癌症在全基因及 miRNA 关系,对比三种癌症散点如何分布并提出下一步思路,通过比较、类聚等方法进行三种癌症的区分。胰腺癌,神经胶质瘤、肾上腺皮质癌利用了 Cytosape 软件生成了三种癌症异常表达网络。这个网络具有由三种关系组成:TF->miRNA 转录因子结合在其识别 DNA 序列后,该基因才开始表达[140];miRNA->target gene 靶基因也叫目的基因,至少 30%人类基因是是 miRNA 的靶基因[141];host gene->miRNA 已经证明宿主基因和 miRNA 具有同向性,miRNA 表达水平受宿主基因控制[142]。还有部分是独立的节点,这些节点不可能是单独存在,一般都是因为研究水平没到位,还有未发现的调控关系。#p#分页标题#e#
..........
总结
本文比较了临床症状比较相似三种遗传性癌症的基因和 miRNAs,利用大数据分析法,以“发生了什么”为起点,利用本文挖掘和人工挖掘相结合的方法入手,挖掘三种癌症的数据,在“可能会发生”的情况下利用随机漫步信任路径算法对 miRNA 数据进行了补充,处理和分析相关数据。通过建立胰腺癌、肾上腺皮质癌、神经胶质瘤三种癌症的数据集。最后分析“为什么发生”和“需要做什么”为指导,采用两种方式相结合的方法建立数学模型。利用 Cytosape 软件构建了三种癌症关于 miRNA、靶基因、转录因子的实验证实调控网络,建立可视化分析网络图,直观的对数据进行分析,两两对比分析建立典型基因发表网络,进行重点分析,得到对比结论。最后通过三种癌症的对比,挖掘出三种相关癌症的一同表达。对比三种癌症的异常表达网络,揭示癌症的发生并不是一个基因单独作用造成的,而是通过 miRNA 与基因相互调节,与蛋白质相互结合,通过异常的表达miRNA 进行错误信息传导,而引起的连锁反应。同时利用类聚观点用已搜集的数据建立miRNA和基因对应关系。采用类聚热图方式,体现三种癌症在全基因及 miRNA 关系,分析了热点分布,相似散点的影响,以此为契机进行三种癌症的临床区分及早期预防。通过聚类热图方式试图通过对比胰腺癌与肾上腺皮质癌、胰腺癌和神经胶质瘤、肾上腺皮质癌和神经胶质瘤两两对比,试图挖掘出各自的发病机理与临床症状之间的关系。在挖掘数据的过程中,我们也会发现数据中过量表达或是低量表达的种种不同,这些解开了生物信息学神秘的面纱,揭示了生物循环的奥秘。
..........
参考文献(略)

