本文是一篇计算机论文,计算机的应用在中国越来越普遍,改革开放以后,中国计算机用户的数量不断攀升,应用水平不断提高,特别是互联网、通信、多媒体等领域的应用取得了不错的成绩。(以上内容来自百度百科)今天为大家推荐一篇计算机理论论文,供大家参考。
第 1 章 绪 论
1.1 本文的研究目的和意义
拟合回归是一类用于分析和确定多种变量之间定量相互依赖关系的算法模型,是在多种学科中被广泛使用的数据挖掘方法。它包括了许多对自变量和因变量之间关系进行建模或者预测的算法与模型,例如:线性回归[1]、人工神经网络[2]、支持向量机[3]、深度学习[4]等。通过这种建立关系模型的方式,它可以用于预测自变量对应的因变量,也可以用于计算自变量对于因变量贡献的大小。拟合回归模型的应用极大地推动了众多学科的深入发展,特别是在生物信息学领域[5][6][7][8][9][10][11][12],拟合回归模型的使用揭示了生物变量之间的关联关系,阐述了关系之中的内在原理。本文研究的目的是通过使用、设计并实现基于拟合回归的计算模型,用于分析生物组学数据,进而回答生物信息学问题,对(1)谷氨酰胺和谷氨酸在人体癌症组织中的代谢特性和(2)特定实验条件下基因间相关性衡量进行了研究,其研究成果具有现实意义:癌症又称恶性肿瘤,是一类由细胞不受控制地快速生长分裂而引起的复杂疾病,具有很强地组织浸润和在不同人体器官之间进行转移的能力[13][14],严重地威胁着人类的生命健康。面对癌症带来的死亡威胁,人类目前最有效的手段是对于癌症的早期诊断和早期治疗,但是其前提条件是对于癌症特征的充分了解与掌握。2011 年,Hanahan 和Weinberg在著名生物学杂志《Cell》上刊文,发表癌症综述的升级版本《Hallmarks of cancer:the next generation》[15]。文中总结了从 2000 年到 2011 年,十一年以来人类在癌症研究领域中的新发现与新热点,并重新概括了癌症的特征,将上一版本[16]中的 6 个癌症特征(如图 1.1-A 所示)扩展成为 10 个(如图 1.1-B 所示),其中新增的癌症特征包括以葡萄糖为代表的能量代谢的异常改变。但是目前人类对于癌症的特征,特别是基于代谢的癌症特征的了解严重不足,不能够有效地减少癌症对于人类生命健康所造成的危害,癌症仍然是造成人类死亡的第二元凶。
..........
1.2 本文的主要工作
本文的两部分主要研究工作不仅都是基于拟合回归的思想,而且恰好分别是拟合回归两种主要用途在具体生物信息学问题中的实际应用。此外,两部分主要研究工作之间存在着内在的应用关系,计划在未来的研究工作中进一步结合。
1. 谷氨酰胺和谷氨酸在多种癌症中的代谢分析
本文(1)使用 11 种癌症和其对照正常组织的转录组学数据研究谷氨酰胺和谷氨酸在癌症组织中的代谢异常,相较于仅使用细胞系实验和动物模型数据,其结果更能够如实地反映谷氨酰胺和谷氨酸代谢在人体癌症组织中的真实情况。(2)采用多元多重线性回归模型,用于计算每种类型的癌症组织中谷氨酰胺/谷氨酸参与 7 种合成代谢过程中产物合成的程度,相较于其它的计算模型,其结果不仅能够对参与程度做出准确的衡量,而且可以提供与参与程度相关的统计显著性,用于发现由癌症而非偶然因素引起的谷氨酰胺和谷氨酸代谢的改变,更具应用性。迄今为止,这是在国内外的众多研究工作中,首次对多种类型癌症组织中谷氨酰胺/谷氨酸参与生物过程水平进行的横向比较。本工作主要针对以下四个问题开展相关研究:(1)癌症组织中谷氨酰胺和谷氨酸的合成和摄入是否显著增强?(2)癌症组织中谷氨酰胺和谷氨酸的基础代谢水平是否显著提高?(3)不同类型的癌症组织中谷氨酰胺和谷氨酸都参与了哪些重要的生物过程,参与水平如何,是否存在替代的生物过程?(4)谷氨酰胺及谷氨酸代谢和癌症组织微环境,特别是与癌症组织中活性氧簇的关系?
.........
第 2 章 生物数据和相关计算方法简介
2.1 生物数据
2.1.1 生物数据概述
生物数据是通过科学技术手段从生物界直接收集或者测量而来的数据。这些数据经过系统地加工、处理和整合,以多种形式存储在全世界不同的数据库中。特别是随着科学技术的日益提高,生物数据的收集和测量进入了“高通量”时代。这些通过“高通量”技术收集而来的生物数据被称为“组学”数据,是现代生物信息学使用和分析数据的重要组成部分。如图 2.1 所示,总结了本文在第一部分主要研究工作:谷氨酰胺和谷氨酸在多种癌症中的代谢分析和在第二部分主要研究工作:基于拟合回归模型(多特征相似性模型)的基因间相关性研究中使用和分析的来自 13 个生物数据库的 4 种组学数据,包括:(1)转录组学数据、(2)基因组学数据、(3)代谢组学数据、(4)蛋白质组学数据。
..........
2.2 线性回归
线性回归(linear regression)是一种机器学习模型,用于描述因变量 y 和自变量x(或者自变量向量)之间的线性关系[1]。其中(1)多元普通线性回归模型是众多线性回归模型的基础,(2)而含有交叉项的多元线性回归模型是在本文的第一部分主要研究工作中使用的模型。因此,下面的章节将重点介绍这两种线性回归模型:可以找到满足条件的超平面有很多,那么选择哪个超平面才最合理?一种朴素而深刻的思路是,这个最合理的超平面应该是可以使得两类样本数据点获得最大间隔的超平面。从概率论的角度而言,选择最大间隔超平面有一个非常直观的优点便是可以使得置信度最小点获得最大的置信度。支持向量机的核心思想便基于此,所以它又被称为最大间隔分类器,其基本原理如图 2.4 所示。事实上求解最大间隔超平面是一个二次凸优化问题。在支持向量机模型中,这个问题使用拉格朗日乘子法求解。下面将通过数学模型描述本文使用的基于线性核函数的线性支持向量机模型。
..........
第 3 章 谷氨酰胺和谷氨酸在多种癌症中的代谢分析.....27
3.1 研究背景 .............. 27
3.2 实验数据与研究方法 .... 29
3.3 实验结果 .............. 34
3.4 结果分析与讨论 ........ 54
3.5 本章小结 .............. 55
第 4 章 基于拟合回归模型的基因间相关性研究.........57
4.1 研究背景 .............. 57
4.2 研究方法 .............. 59
4.3 结果比较方法 .......... 71
4.4 实验结果 .............. 76
4.5 结果分析与讨论 ........ 86
4.6 本章小结 .............. 88
第 5 章 总结与展望...........91
第 4 章 基于拟合回归模型的基因间相关性研究
4.1 研究背景
如何衡量特定实验条件下的基因间相关性是一个重要的基础性课题,并且在生物信息学领域仍然是一项具有挑战性的工作。在细胞环境或者生物通路中,基因及其蛋白质产物并不是单独地行使作用,而是相互协作完成各种繁复的生物功能。并且在不同的环境中,基因间的协作关系也千差万别。为了探究目标基因之间的内在联系,准确地衡量特定实验条件下的基因间相关性就变得愈加重要。例如,被衡量的基因间相关性可以作为基因与基因之间连接的权重,在构建特定实验条件下的生物网络时发挥作用[47][48][49][50][51];也可以用于预测在某种环境中的基因新功能[52][53][54]。本章研究的重点是拟设计并实现基于拟合回归的算法模型,用于准确地衡量特定实验条件下的基因间相关性。在以往的国内外研究中,度量特定实验条件下的基因间相关性,首先收集基于这个实验条件的基因表达数据,然后使用共表达相似性来衡量。共表达相似性,通过使用两个基因在大量样本中表达模式的相似性来衡量特定实验条件下的基因间相关性,主要使用共表达分析算法进行计算[55][56][57][58],例如:皮尔森相关系数(Pearson correlation coefficient,PCC)[59]、斯皮尔曼相关系数(Spearman rank correlation,SRC)[60]、互信息(mutual information,MI)[61][62][63][64]、皮尔森偏相关系数(partial Pearsoncorrelation,PPC)[65][66][67]、条件互信息(conditional mutual information,CMI)[68]等。使用这些算法分析大量基因表达数据,生物学家们建立了众多的基因共表达数据库,例如:COXPRESdb[69]和 GeneFriends[70]数据库。与此同时,国内外大量的研究也表明,先验知识相似性通过衡量两个基因的先验知识,例如功能注释的相似性,也可以用于衡量基因之间的相关性[71][72][73]。其中用于计算相似性的先验知识来源于大量的公共数据库,例如:基因功能数据库 Gene Ontology(GO)[74];生物通路数据库 Kyoto Encyclopedia ofGenes and Genomes(KEGG)[75]、Reactome[76];同源基因数据库 OrthoDB[77];蛋白质间相互作用数据库 Human Protein Reference Database(HPRD)[78]、Database of InteractingProteins(DIP)[79]、Negatome[80];转录调控数据库 Human Transcriptional RegulationInteractions database(HTRIdb)[81]、Transcriptional Regulatory Relationships Unravelled bySentence-based Text-mining(TRRUST)[82]。#p#分页标题#e#
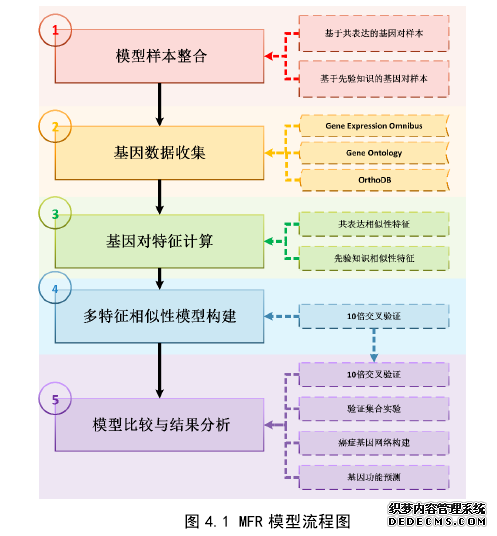
.........
总结
拟合回归是一类用于分析和确定多种变量之间定量相互依赖关系的算法模型,是在多种学科中被广泛使用的数据挖掘方法。它的应用极大地推动了众多学科的深入发展。特别是在生物信息学领域,拟合回归模型的使用揭示了生物变量之间的关联关系,阐述了关系之中的内在原理。本文使用、设计并实现基于拟合回归的算法模型,用于分析生物组学数据,进而回答生物信息学问题。本文的主要研究工作集中在如下两方面:本文(1)使用 11 种癌症和其对照正常组织的转录组学数据研究谷氨酰胺和谷氨酸在癌症组织中的代谢异常,其结果更能够如实地反映谷氨酰胺和谷氨酸代谢在人体癌症组织中的真实情况。(2)采用多元多重线性回归模型,用于计算每种类型的癌症组织中谷氨酰胺/谷氨酸参与 7 种合成代谢过程中产物合成的程度及其统计显著性。迄今为止,这是在国内外的众多研究工作中,首次对多种类型癌症组织中谷氨酰胺/谷氨酸参与生物过程水平进行的横向比较。通过系统的分析,对谷氨酰胺和谷氨酸在癌症组织中参与生物过程的情况有了全新的认识。发现谷氨酰胺和谷氨酸在不同的癌症组织中参与 7 种合成代谢过程的程度存在显著异常。这些异常,尤其是其中谷氨酰胺和谷氨酸参与某些合成过程显著增强可以作为基于谷氨酰胺和谷氨酸代谢的癌症新特征。虽然这些新的发现还需要进一步的生物实验验证,但是可以为以谷氨酰胺和谷氨酸代谢为靶点的癌症治疗手段提供新思路与理论依据。
..........
参考文献(略)

